

Introducing the Shell¶
Questions:
What is a command shell and why would I use one?
Objectives:
Explain how the shell relates to the keyboard, the screen, the operating system, and users’ programs.
Explain when and why command-line interfaces should be used instead of graphical interfaces.
Keypoints:
Explain the steps in the shell’s read-run-print cycle.
Most commands take options (flags) which begin with a
-.Identify the actual command, options, and filenames in a command-line call.
Demonstrate the use of tab completion and explain its advantages.
A shell is a program whose primary purpose is to read commands and run other programs.
The shell’s main advantages are its high action-to-keystroke ratio, its support for automating repetitive tasks, and its capacity to access networked machines.
The shell’s main disadvantages are its primarily textual nature and how cryptic its commands and operation can be.
Background¶
Humans and computers commonly interact in many different ways, such as through a keyboard and mouse, touch screen interfaces, or using speech recognition systems. The most widely used way to interact with personal computers is called a graphical user interface (GUI). With a GUI, we give instructions by clicking a mouse and using menu-driven interactions.
While the visual aid of a GUI makes it intuitive to learn, this way of delivering instructions to a computer scales very poorly. Imagine the following task: for a literature search, you have to copy the third line of one thousand text files in one thousand different directories and paste it into a single file. Using a GUI, you would not only be clicking at your desk for several hours, but you could potentially also commit an error in the process of completing this repetitive task. This is where we take advantage of the Unix shell. The Unix shell is both a command-line interface (CLI) and a scripting language, allowing such repetitive tasks to be done automatically and fast. With the proper commands, the shell can repeat tasks with or without some modification as many times as we want. Using the shell, the task in the literature example can be accomplished in seconds.
The Shell:
The shell is a program where users can type commands. With the shell, it’s possible to invoke complicated programs like climate modeling software or simple commands that create an empty directory with only one line of code. The most popular Unix shell is Bash (the Bourne Again SHell — so-called because it’s derived from a shell written by Stephen Bourne). Bash is the default shell on most modern implementations of Unix and in most packages that provide Unix-like tools for Windows.
Using the shell will take some effort and some time to learn. While a GUI presents you with choices to select, CLI choices are not automatically presented to you, so you must learn a few commands like new vocabulary in a language you’re studying. However, unlike a spoken language, a small number of “words” (i.e. commands) gets you a long way, and we’ll cover those essential few today.
The grammar of a shell allows you to combine existing tools into powerful pipelines and handle large volumes of data automatically. Sequences of commands can be written into a script, improving the reproducibility of workflows.
In addition, the command line is often the easiest way to interact with remote machines and supercomputers. Familiarity with the shell is near essential to run a variety of specialized tools and resources including high-performance computing systems. As clusters and cloud computing systems become more popular for scientific data crunching, being able to interact with the shell is becoming a necessary skill. We can build on the command-line skills covered here to tackle a wide range of scientific questions and computational challenges.
Getting Started¶
Opening The Shell Via CyVerse¶
Follow along with Tuesday’s demo to start the RStudio environment with the input for this lesson.
For your RStudio instance, set the input folder to /iplant/home/shared/ag2pi_workshop
The login screen should look like this:
And we will be using the shell inside RStudio for these lessons.
When the shell is first opened, you are presented with a prompt, indicating that the shell is waiting for input.
$
The shell typically uses $ as the prompt, but may use a different symbol.
In the examples for this lesson, we’ll show the prompt as $.
Most importantly:
when typing commands, either from these lessons or from other sources,
do not type the prompt, only the commands that follow it.
So let’s try our first command, ls which is short for listing.
This command will list the contents of the current directory:
$ ls
input kitematic
Command not found¶
If the shell can’t find a program whose name is the command you typed, it will print an error message such as:
$ ks
ks: command not found
This might happen if the command was mis-typed or if the program corresponding to that command is not installed.
Navigating Files and Directories¶
Questions:
How can I move around on my computer?
How can I see what files and directories I have?
How can I specify the location of a file or directory on my computer?
Objectives:
Explain the similarities and differences between a file and a directory.
Translate an absolute path into a relative path and vice versa.
Construct absolute and relative paths that identify specific files and directories.
Use options and arguments to change the behaviour of a shell command
Demonstrate the use of tab completion, and explain its advantages.
Keypoints:
The file system is responsible for managing information on the disk.
Information is stored in files, which are stored in directories (folders).
Directories can also store other directories, which forms a directory tree.
cd [path]changes the current working directory.ls [path]prints a listing of a specific file or directory;lson its own lists the current working directory.pwdprints the user’s current working directory./on its own is the root directory of the whole file system.A relative path specifies a location starting from the current location.
An absolute path specifies a location from the root of the file system.
Directory names in a path are separated with
/on Unix, but\\on Windows...means ‘the directory above the current one’;.on its own means ‘the current directory’.
The File System¶
The part of the operating system responsible for managing files and directories is called the file system. It organizes our data into files, which hold information, and directories (also called ‘folders’), which hold files or other directories.
Several commands are frequently used to create, inspect, rename, and delete files and directories. To start exploring them, we’ll go to our open shell window.
First let’s find out where we are by running a command called pwd
(which stands for ‘print working directory’). Directories are like places - at any time
while we are using the shell we are in exactly one place, called
our current working directory. Commands mostly read and write files in the
current working directory, i.e. ‘here’, so knowing where you are before running
a command is important. pwd shows you where you are:
$ pwd
/home/rstudio
Here,
the computer's response is ``/home/rstudio``\ ,
which is the application's **home directory**\ :
Home Directory Variation¶
The home directory path will look different on different operating systems.
On Linux it may look like /home/nelle,
and on Windows it will be similar to C:\Documents and Settings\nelle or
C:\Users\nelle.
(Note that it may look slightly different for different versions of Windows.)
In future examples, we’ve used Mac output as the default - Linux and Windows
output may differ slightly, but should be generally similar.
To understand what a ‘home directory’ is, let’s have a look at how the file system as a whole is organized. For the sake of this example, we’ll be illustrating the filesystem inside our CyVerse app. After this illustration, you’ll be learning commands to explore your own filesystem, which will be constructed in a similar way, but not be exactly identical.
On the CyVerse app, the filesystem is quite complex.
An example filesystem diagram might look like this:
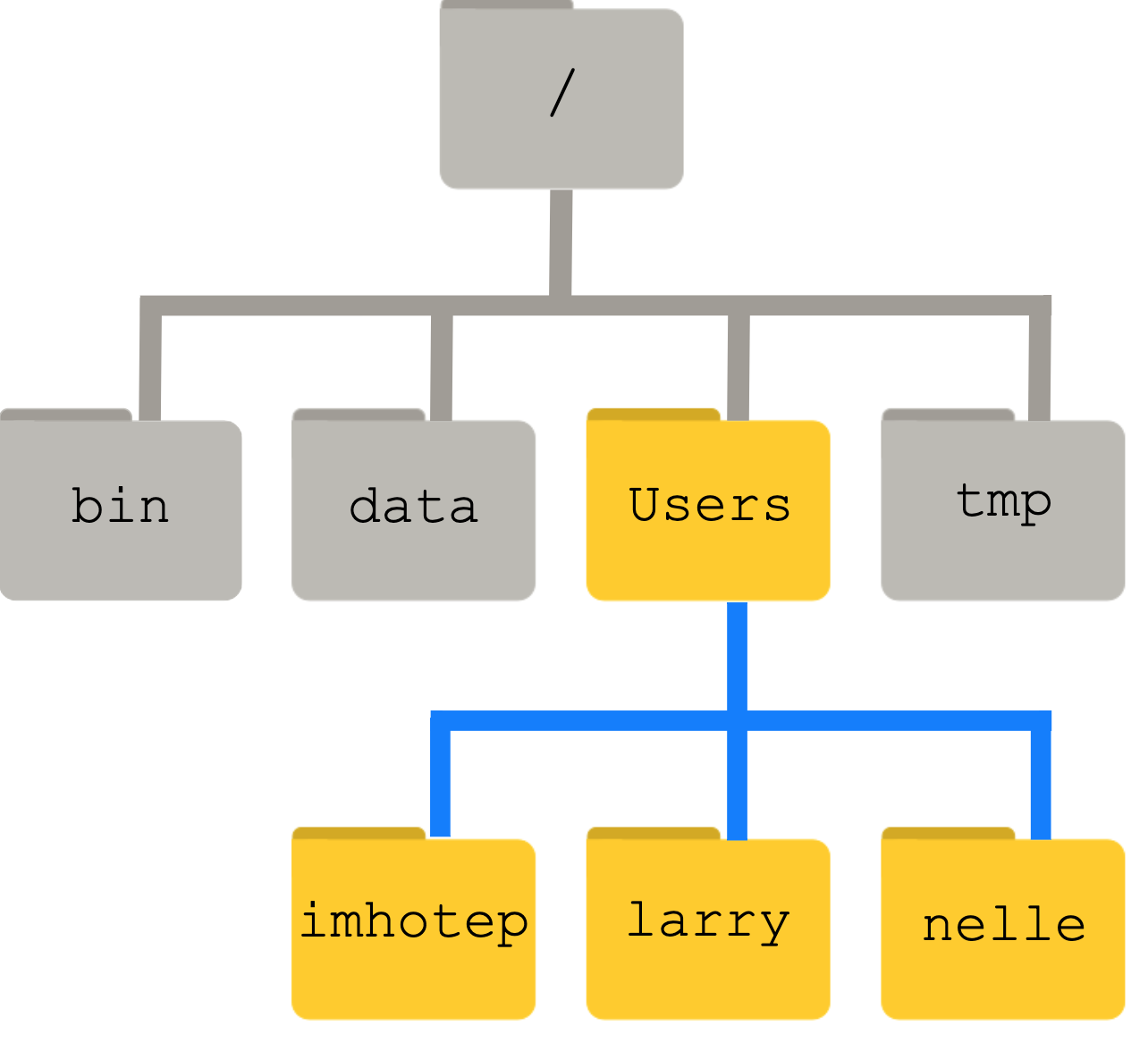
At the top is the root directory
that holds everything else.
We refer to it using a slash character, /, on its own;
this is the leading slash in /home/rstudio.
Inside that directory are several other directories:
bin (which is where some built-in programs are stored),
data (for miscellaneous data files),
Users (where users’ personal directories are located),
tmp (for temporary files that don’t need to be stored long-term),
and so on.
We know that our current working directory /home/rstudio is stored inside /home
because /home is the first part of its name.
Similarly,
we know that /home is stored inside the root directory /
because its name begins with /.
Slashes¶
Notice that there are two meanings for the
/character. When it appears at the front of a file or directory name, it refers to the root directory. When it appears inside a path, it’s just a separator.
Now let’s learn the command that will let us see the contents of our
own filesystem. We can see what’s in our home directory by running ls:
$ ls
/home/rstudio
(Again, your results may be slightly different running these commands on a different computer, depending on your operating system and how you have customized your filesystem.)
lsprints the names of the files and directories in the current directory. We can make its output more comprehensible by using the-Foption (also known as a switch or a flag) , which tellslsto classify the output by adding a marker to file and directory names to indicate what they are:
a trailing
/indicates that this is a directory@indicates a link*indicates an executableDepending on your default options, the shell might also use colors to indicate whether each entry is a file or directory.
$ ls -F
input/ kitematic/
Clearing your terminal¶
If your screen gets too cluttered, you can clear your terminal using the
clearcommand. You can still access previous commands using ↑ and ↓ to move line-by-line, or by scrolling in your terminal.Here, we can see that our home directory contains only sub-directories. Any names in your output that don’t have a classification symbol, are plain old files.
General syntax of a shell command¶
Consider the command below as a general example of a command, which we will dissect into its component parts:
$ ls -F /
ls is the command, with an option -F and an argument /.
We’ve already encountered options (also called switches or flags) which
either start with a single dash (-) or two dashes (--), and they change the behavior of a command.
Arguments tell the command what to operate on (e.g. files and directories).
Sometimes options and arguments are referred to as parameters.
A command can be called with more than one option and more than one argument: but a
command doesn’t always require an argument or an option.
Each part is separated by spaces: if you omit the space
between ls and -F the shell will look for a command called ls-F, which
doesn’t exist. Also, capitalization can be important. For example, ls -s will display the size of files and directories alongside the names, while ls -S will sort the files and directories by size, as shown below:
$ ls -s input/ag2pi_workshop/data
total 124
4 amino-acids.txt 4 animals.txt 4 elements 4 morse.txt 12 planets.txt 76 sunspot.txt
0 animal-counts 4 basilisk.dat 4 minotaur.dat 4 pdb 4 salmon.txt 4 unicorn.dat
$ ls -S input/ag2pi_workshop/data
sunspot.txt elements basilisk.dat minotaur.dat amino-acids.txt salmon.txt
planets.txt pdb unicorn.dat morse.txt animals.txt animal-counts
Putting all that together, our command above gives us a listing
of files and directories in the root directory /.
An example of the output you might get from the RStudio app on CyVerse with the above command is given below:
$ ls -F /
bin/ home/ lib32/ mnt/ root/ sys/
boot/ init* lib64/ nginx.conf.tmpl run/ tmp/
dev/ irods-icommands-4.1.10-ubuntu14-x86_64.deb libexec/ opt/ sbin/ usr/
etc/ lib/ media/ proc/ srv/ var/
Getting help¶
lshas lots of other options. There are two common ways to find out how to use a command and what options it accepts:
We can pass a
--helpoption to the command, such as: .. code-block:$ ls --help
We can read its manual with
man, such as: .. code-block:$ man ls
Depending on your environment you might find that only one of these works (either ``man`` or ``–help``, eg. ``man`` works for macOS and ``–help`` typically works for Git Bash).
We’ll describe both ways below.
The --help option¶
Many bash commands, and programs that people have written that can be run from within bash, support a
--helpoption to display more information on how to use the command or program.
$ ls --help
Usage: ls [OPTION]... [FILE]...
List information about the FILEs (the current directory by default).
Sort entries alphabetically if none of -cftuvSUX nor --sort is specified.
Mandatory arguments to long options are mandatory for short options too.
-a, --all do not ignore entries starting with .
-A, --almost-all do not list implied . and ..
--author with -l, print the author of each file
-b, --escape print C-style escapes for nongraphic characters
--block-size=SIZE scale sizes by SIZE before printing them; e.g.,
'--block-size=M' prints sizes in units of
1,048,576 bytes; see SIZE format below
-B, --ignore-backups do not list implied entries ending with ~
-c with -lt: sort by, and show, ctime (time of last
modification of file status information);
with -l: show ctime and sort by name;
otherwise: sort by ctime, newest first
-C list entries by columns
--color[=WHEN] colorize the output; WHEN can be 'always' (default
if omitted), 'auto', or 'never'; more info below
-d, --directory list directories themselves, not their contents
-D, --dired generate output designed for Emacs' dired mode
-f do not sort, enable -aU, disable -ls --color
-F, --classify append indicator (one of */=>@|) to entries
--file-type likewise, except do not append '*'
--format=WORD across -x, commas -m, horizontal -x, long -l,
single-column -1, verbose -l, vertical -C
--full-time like -l --time-style=full-iso
-g like -l, but do not list owner
--group-directories-first
group directories before files;
can be augmented with a --sort option, but any
use of --sort=none (-U) disables grouping
-G, --no-group in a long listing, don't print group names
-h, --human-readable with -l and/or -s, print human readable sizes
(e.g., 1K 234M 2G)
--si likewise, but use powers of 1000 not 1024
-H, --dereference-command-line
follow symbolic links listed on the command line
--dereference-command-line-symlink-to-dir
follow each command line symbolic link
that points to a directory
--hide=PATTERN do not list implied entries matching shell PATTERN
(overridden by -a or -A)
--indicator-style=WORD append indicator with style WORD to entry names:
none (default), slash (-p),
file-type (--file-type), classify (-F)
-i, --inode print the index number of each file
-I, --ignore=PATTERN do not list implied entries matching shell PATTERN
-k, --kibibytes default to 1024-byte blocks for disk usage
-l use a long listing format
-L, --dereference when showing file information for a symbolic
link, show information for the file the link
references rather than for the link itself
-m fill width with a comma separated list of entries
-n, --numeric-uid-gid like -l, but list numeric user and group IDs
-N, --literal print raw entry names (don't treat e.g. control
characters specially)
-o like -l, but do not list group information
-p, --indicator-style=slash
append / indicator to directories
-q, --hide-control-chars print ? instead of nongraphic characters
--show-control-chars show nongraphic characters as-is (the default,
unless program is 'ls' and output is a terminal)
-Q, --quote-name enclose entry names in double quotes
--quoting-style=WORD use quoting style WORD for entry names:
literal, locale, shell, shell-always,
shell-escape, shell-escape-always, c, escape
-r, --reverse reverse order while sorting
-R, --recursive list subdirectories recursively
-s, --size print the allocated size of each file, in blocks
-S sort by file size, largest first
--sort=WORD sort by WORD instead of name: none (-U), size (-S),
time (-t), version (-v), extension (-X)
--time=WORD with -l, show time as WORD instead of default
modification time: atime or access or use (-u);
ctime or status (-c); also use specified time
as sort key if --sort=time (newest first)
--time-style=STYLE with -l, show times using style STYLE:
full-iso, long-iso, iso, locale, or +FORMAT;
FORMAT is interpreted like in 'date'; if FORMAT
is FORMAT1<newline>FORMAT2, then FORMAT1 applies
to non-recent files and FORMAT2 to recent files;
if STYLE is prefixed with 'posix-', STYLE
takes effect only outside the POSIX locale
-t sort by modification time, newest first
-T, --tabsize=COLS assume tab stops at each COLS instead of 8
-u with -lt: sort by, and show, access time;
with -l: show access time and sort by name;
otherwise: sort by access time, newest first
-U do not sort; list entries in directory order
-v natural sort of (version) numbers within text
-w, --width=COLS set output width to COLS. 0 means no limit
-x list entries by lines instead of by columns
-X sort alphabetically by entry extension
-Z, --context print any security context of each file
-1 list one file per line. Avoid '\n' with -q or -b
--help display this help and exit
--version output version information and exit
The SIZE argument is an integer and optional unit (example: 10K is 10*1024).
Units are K,M,G,T,P,E,Z,Y (powers of 1024) or KB,MB,... (powers of 1000).
Using color to distinguish file types is disabled both by default and
with --color=never. With --color=auto, ls emits color codes only when
standard output is connected to a terminal. The LS_COLORS environment
variable can change the settings. Use the dircolors command to set it.
Exit status:
0 if OK,
1 if minor problems (e.g., cannot access subdirectory),
2 if serious trouble (e.g., cannot access command-line argument).
GNU coreutils online help: <http://www.gnu.org/software/coreutils/>
Full documentation at: <http://www.gnu.org/software/coreutils/ls>
or available locally via: info '(coreutils) ls invocation'
Unsupported command-line options¶
If you try to use an option (flag) that is not supported,
lsand other commands will usually print an error message similar to:
$ ls -j
ls: invalid option -- 'j'
Try 'ls --help' for more information.
The man command¶
man is short for manual, which will open the unix manual in your terminal window.
So, the other way to learn about
lsis to type
$ man ls
This will turn your terminal into a page with a description
of the ls command and its options.
To navigate through the man pages,
you may use ↑ and ↓ to move line-by-line,
or try B and Spacebar``Spacebar to skip up and down by a full page.
To search for a character or word in the `man` pages,
use ``/ followed by the character or word you are searching for.
Sometimes a search will result in multiple hits. If so, you can move between hits using N (for moving forward) and Shift + N (for moving backward).
To quit the man pages, press Q.
Manual pages on the web¶
Of course there is a third way to access help for commands: searching the internet via your web browser. When using internet search, including the phrase
unix man pagein your search query will help to find relevant results.GNU provides links to its manuals including the core GNU utilities, which covers many commands introduced within this lesson.
Exploring More ls Flags¶
You can also use two options at the same time. What does the command
lsdo when used with the-loption? What about if you use both the-land the-hoption?Some of its output is about properties that we do not cover in this lesson (such as file permissions and ownership), but the rest should be useful nevertheless.
Solution
The -l option makes ls use a long listing format, showing not only
the file/directory names but also additional information such as the file size
and the time of its last modification. If you use both the -h option and the -l option,
this makes the file size ‘human readable’, i.e. displaying something like 5.3K
instead of 5369.
Listing in Reverse Chronological Order¶
By default
lslists the contents of a directory in alphabetical order by name. The commandls -tlists items by time of last change instead of alphabetically. The commandls -rlists the contents of a directory in reverse order. Which file is displayed last when you combine the-tand-rflags? Hint: You may need to use the-lflag to see the last changed dates.
Solution
The most recently changed file is listed last when using -rt. This
can be very useful for finding your most recent edits or checking to
see if a new output file was written.
Exploring Other Directories¶
Not only can we use
lson the current working directory, but we can use it to list the contents of a different directory. Let’s take a look at ourag2pi_workshopdirectory by runningls -F input/ag2pi_workshop, i.e., the commandlswith the-Foption and the argumentinput/ag2pi_workshop. The argumentinput/ag2pi_workshoptellslsthat we want a listing of something other than our current working directory:
$ ls -F input/ag2pi_workshop
ag-data/ creatures/ data/ north-pacific-gyre/ notes.txt pizza.cfg solar.pdf writing/
Your output should be a list of all the files and sub-directories in the ag2pi_workshop directory. On many systems, the command line Desktop directory is the same as your GUI Desktop. Take a look at your Desktop to confirm that your output is accurate.
As you may now see, using a bash shell is strongly dependent on the idea that your files are organized in a hierarchical file system. Organizing things hierarchically in this way helps us keep track of our work: it’s possible to put hundreds of files in our home directory, just as it’s possible to pile hundreds of printed papers on our desk, but it’s a self-defeating strategy.
Now that we know the ag2pi_workshop directory is located in our input directory, we
can do two things.
First, we can look at its contents, using the same strategy as before, passing
a directory name to ls:
$ ls -F input/ag2pi_workshop
ag-data/ creatures/ data/ north-pacific-gyre/ notes.txt pizza.cfg solar.pdf writing/
Second, we can actually change our location to a different directory, so we are no longer located in our home directory.
The command to change locations is cd followed by a
directory name to change our working directory.
cd stands for ‘change directory’,
which is a bit misleading:
the command doesn’t change the directory,
it changes the shell’s idea of what directory we are in.
The cd command is akin to double clicking a folder in a graphical interface to get into a folder.
Let’s say we want to move to the data directory we saw above. We can
use the following series of commands to get there:
$ cd input
$ cd ag2pi_workshop
$ cd data
These commands will move us from our home directory into our input directory, then into
the ag2pi_workshop directory, then into the data directory. You will notice that cd doesn’t print anything. This is normal. Many shell commands will not output anything to the screen when successfully executed. But if we run pwd after it, we can see that we are now
in /home/rstudio/input/ag2pi_workshop/data.
If we run ls -F without arguments now,
it lists the contents of /home/rstudio/input/ag2pi_workshop/data,
because that’s where we now are:
$ pwd
/home/rstudio/input/ag2pi_workshop/data
$ ls -F
amino-acids.txt animals.txt elements/ morse.txt planets.txt sunspot.txt
animal-counts/ basilisk.dat minotaur.dat pdb/ salmon.txt unicorn.dat
We now know how to go down the directory tree (i.e. how to go into a subdirectory) but how do we go up (i.e. how do we leave a directory and go into its parent directory)? We might try the following:
$ cd ag2pi_workshop
-bash: cd: ag2pi_workshop: No such file or directory
But we get an error! Why is this?
With our methods so far,
cd can only see sub-directories inside your current directory. There are
different ways to see directories above your current location; we’ll start
with the simplest.
There is a shortcut in the shell to move up one directory level that looks like this:
$ cd ..
.. is a special directory name meaning
“the directory containing this one”,
or more succinctly,
the parent of the current directory.
Sure enough,
if we run pwd after running cd .., we’re back in /home/rstudio/input/ag2pi_workshop:
$ pwd
/home/rstudio/input/ag2pi_workshop
The special directory .. doesn’t usually show up when we run ls. If we want
to display it, we can add the -a option to ls -F:
$ ls -F -a
./ ../ ag-data/ creatures/ data/ north-pacific-gyre/ notes.txt pizza.cfg solar.pdf writing/
-a stands for ‘show all’;
it forces ls to show us file and directory names that begin with .,
such as .. (which, if we’re in /home/rstudio, refers to the /home directory)
As you can see,
it also displays another special directory that’s just called .,
which means ‘the current working directory’.
It may seem redundant to have a name for it,
but we’ll see some uses for it soon.
Note that in most command line tools, multiple options can be combined
with a single - and no spaces between the options: ls -F -a is
equivalent to ls -Fa.
Orthogonality¶
The special names
.and..don’t belong tocd; they are interpreted the same way by every program. For example, if we are in/home/rstudio/input/ag2pi_workshop, the commandls ..will give us a listing of/home/rstudio/input. When the meanings of the parts are the same no matter how they’re combined, programmers say they are orthogonal: Orthogonal systems tend to be easier for people to learn because there are fewer special cases and exceptions to keep track of.These then, are the basic commands for navigating the filesystem on your computer:
pwd,lsandcd. Let’s explore some variations on those commands. What happens if you typecdon its own, without giving a directory?
$ cd
How can you check what happened? pwd gives us the answer!
$ pwd
/home/rstudio
It turns out that cd without an argument will return you to your home directory,
which is great if you’ve gotten lost in your own filesystem.
Let’s try returning to the data directory from before. Last time, we used
three commands, but we can actually string together the list of directories
to move to data in one step:
$ cd input/ag2pi_workshop/data
Check that we’ve moved to the right place by running pwd and ls -F
If we want to move up one level from the data directory, we could use cd ... But
there is another way to move to any directory, regardless of your
current location.
So far, when specifying directory names, or even a directory path (as above),
we have been using relative paths. When you use a relative path with a command
like ls or cd, it tries to find that location from where we are,
rather than from the root of the file system.
However, it is possible to specify the absolute path to a directory by
including its entire path from the root directory, which is indicated by a
leading slash. The leading / tells the computer to follow the path from
the root of the file system, so it always refers to exactly one directory,
no matter where we are when we run the command.
This allows us to move to our ag2pi_workshop directory from anywhere on
the filesystem (including from inside data). To find the absolute path
we’re looking for, we can use pwd and then extract the piece we need
to move to ag2pi_workshop.
$ pwd
/home/rstudio/input/ag2pi_workshop
$ cd /home/rstudio/input/ag2pi_workshop
Run pwd and ls -F to ensure that we’re in the directory we expect.
Two More Shortcuts¶
The shell interprets the character
~(tilde) at the start of a path to mean “the current user’s home directory”. For example, if Nelle’s home directory is/Users/nelle, then~/datais equivalent to/Users/nelle/data. This only works if it is the first character in the path:here/there/~/elsewhereis nothere/there/Users/nelle/elsewhere.Another shortcut is the
-(dash) character.cdwill translate-into the previous directory I was in, which is faster than having to remember, then type, the full path. This is a very efficient way of moving back and forth between directories. The difference betweencd ..andcd -is that the former brings you up, while the latter brings you back. You can think of it as the Last Channel button on a TV remote.
Absolute vs Relative Paths¶
Starting from
/Users/amanda/data, which of the following commands could Amanda use to navigate to her home directory, which is/Users/amanda?
cd .cd /cd /home/amandacd ../..cd ~cd homecd ~/data/..cdcd ..
Solution
No:
.stands for the current directory.No:
/stands for the root directory.No: Amanda’s home directory is
/Users/amanda.No: this goes up two levels, i.e. ends in
/Users.Yes:
~stands for the user’s home directory, in this case/Users/amanda.No: this would navigate into a directory
homein the current directory if it exists.Yes: unnecessarily complicated, but correct.
Yes: shortcut to go back to the user’s home directory.
Yes: goes up one level.
Relative Path Resolution¶
Using the filesystem diagram below, if
pwddisplays/Users/thing, what willls -F ../backupdisplay?
../backup: No such file or directory2012-12-01 2013-01-08 2013-01-272012-12-01/ 2013-01-08/ 2013-01-27/original/ pnas_final/ pnas_sub/
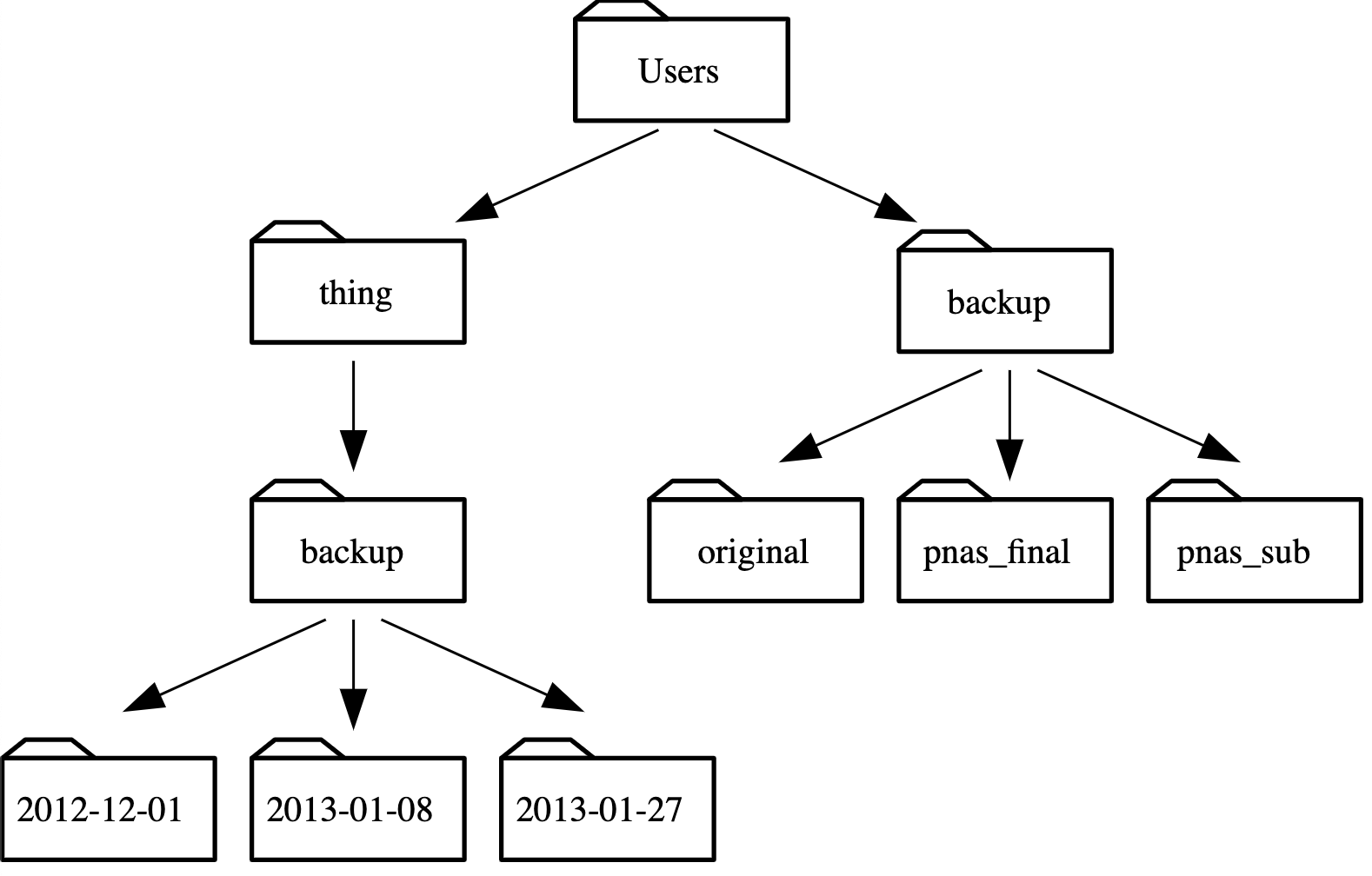
Solution
No: there is a directory
backupin/Users.- No: this is the content of
Users/thing/backup, but with
..we asked for one level further up.
- No: this is the content of
No: see previous explanation.
Yes:
../backup/refers to/Users/backup/.
ls Reading Comprehension¶
Using the filesystem diagram below, if
pwddisplays/Users/backup, and-rtellslsto display things in reverse order, what command(s) will result in the following output:
pnas_sub/ pnas_final/ original/

ls pwdls -r -Fls -r -F /Users/backup
Solution
No:
pwdis not the name of a directory.Yes:
lswithout directory argument lists files and directories in the current directory.Yes: uses the absolute path explicitly.
Challenge - Nelle’s Pipeline: Organizing Files¶
Knowing this much about files and directories, Nelle is ready to organize the files that the protein assay machine will create. First, she creates a directory called
north-pacific-gyre(to remind herself where the data came from). Inside that, she creates a directory called2012-07-03, which is the date she started processing the samples. She used to use names likeconference-paperandrevised-results, but she found them hard to understand after a couple of years. (The final straw was when she found herself creating a directory calledrevised-revised-results-3.)
Sorting Output¶
Nelle names her directories ‘year-month-day’, with leading zeroes for months and days, because the shell displays file and directory names in alphabetical order. If she used month names, December would come before July; if she didn’t use leading zeroes, November (‘11’) would come before July (‘7’). Similarly, putting the year first means that June 2012 will come before June 2013.
Each of her physical samples is labelled according to her lab’s convention with a unique ten-character ID, such as ‘NENE01729A’. This is what she used in her collection log to record the location, time, depth, and other characteristics of the sample, so she decides to use it as part of each data file’s name. Since the assay machine’s output is plain text, she will call her files
NENE01729A.txt,NENE01812A.txt, and so on. All 1520 files will go into the same directory.Now in her current directory
ag2pi_workshop, Nelle can see what files she has using the command:
$ ls north-pacific-gyre/2012-07-03/
This is a lot to type, but she can let the shell do most of the work through what is called tab completion. If she types:
$ ls nor
and then presses Tab (the tab key on her keyboard),
The shell automatically completes the directory name for her:
$ ls north-pacific-gyre/
If she presses Tab again,
Bash will add 2012-07-03/ to the command,
since it’s the only possible completion.
Pressing Tab again does nothing,
since there are 19 possibilities;
pressing Tab twice brings up a list of all the files,
and so on.
This is called tab completion,
and we will see it in many other tools as we go on.
Working With Files and Directories¶
Questions:
How can I create, copy, and delete files and directories?
How can I edit files?
Objectives:
Create a directory hierarchy that matches a given diagram.
Create files in that hierarchy using an editor or by copying and renaming existing files.
Delete, copy and move specified files and/or directories.
Keypoints:
cp [old] [new]copies a file.mkdir [path]creates a new directory.mv [old] [new]moves (renames) a file or directory.rm [path]removes (deletes) a file.*matches zero or more characters in a filename, so*.txtmatches all files ending in.txt.?matches any single character in a filename, so?.txtmatchesa.txtbut notany.txt.Use of the Control key may be described in many ways, including
Ctrl-X,Control-X, and^X.The shell does not have a trash bin: once something is deleted, it’s really gone.
Most files’ names are
something.extension. The extension isn’t required, and doesn’t guarantee anything, but is normally used to indicate the type of data in the file.Depending on the type of work you do, you may need a more powerful text editor than Nano.
Creating Directories¶
We now know how to explore files and directories, but how do we create them in the first place?
Step one: see where we are and what we already have¶
Let’s go back to our
ag2pi_workshopdirectory in theinputdirectory and usels -Fto see what it contains:
$ pwd
/home/rstudio/input/ag2pi_workshop
$ ls -F
ag-data/ creatures/ data/ north-pacific-gyre/ notes.txt pizza.cfg solar.pdf writing/
Step two: Create a directory¶
Let’s create a new directory called
thesisusing the commandmkdir thesis(which has no output):
$ mkdir thesis
As you might guess from its name,
mkdir means ‘make directory’.
Since thesis is a relative path
(i.e., does not have a leading slash, like /what/ever/thesis),
the new directory is created in the current working directory:
$ ls -F
creatures/ data/ molecules/ north-pacific-gyre/ notes.txt pizza.cfg solar.pdf thesis/ writing/
Since we’ve just created the thesis directory, there’s nothing in it yet:
$ ls -F thesis
Note that mkdir is not limited to creating single directories one at a time. The -p option allows mkdir to create a directory with any number of nested subdirectories in a single operation:
$ mkdir -p thesis/chapter_1/section_1/subsection_1
The -R option to the ls command will list all nested subdirectories wtihin a directory. Let’s use ls -FR to recursively list the new directory hierarchy we just created beneath the thesis directory:
$ ls -FR thesis
chapter_1/
thesis/chapter_1:
section_1/
thesis/chapter_1/section_1:
subsection_1/
thesis/chapter_1/section_1/subsection_1:
Two ways of doing the same thing¶
Using the shell to create a directory is no different than using a file explorer. If you open the current directory using your operating system’s graphical file explorer, the
thesisdirectory will appear there too. While the shell and the file explorer are two different ways of interacting with the files, the files and directories themselves are the same.
Good names for files and directories¶
Complicated names of files and directories can make your life painful when working on the command line. Here we provide a few useful tips for the names of your files.
Don’t use spaces.
Spaces can make a name more meaningful, but since spaces are used to separate arguments on the command line it is better to avoid them in names of files and directories. You can use
-or_instead (e.g.north-pacific-gyre/rather thannorth pacific gyre/).Don’t begin the name with
-(dash).Commands treat names starting with
-as options.Stick with letters, numbers,
.(period or ‘full stop’),-(dash) and_(underscore).Many other characters have special meanings on the command line. We will learn about some of these during this lesson. There are special characters that can cause your command to not work as expected and can even result in data loss.
If you need to refer to names of files or directories that have spaces or other special characters, you should surround the name in quotes (
"").
Create a text file¶
Let’s change our working directory to
thesisusingcd, then run a text editor called Nano to create a file calleddraft.txt:
$ cd thesis
$ nano draft.txt
Which Editor?¶
When we say, ‘
nanois a text editor’ we really do mean ‘text’: it can only work with plain character data, not tables, images, or any other human-friendly media. We use it in examples because it is one of the least complex text editors. However, because of this trait, it may not be powerful enough or flexible enough for the work you need to do after this workshop. On Unix systems (such as Linux and macOS), many programmers use Emacs or Vim (both of which require more time to learn), or a graphical editor such as Gedit. On Windows, you may wish to use Notepad++. Windows also has a built-in editor callednotepadthat can be run from the command line in the same way asnanofor the purposes of this lesson.No matter what editor you use, you will need to know where it searches for and saves files. If you start it from the shell, it will (probably) use your current working directory as its default location. If you use your computer’s start menu, it may want to save files in your desktop or documents directory instead. You can change this by navigating to another directory the first time you ‘Save As…’
Let’s type in a few lines of text. Once we’re happy with our text, we can press
Ctrl+O(press theControlkey and, while holding it down, press theO) to write our data to disk (we’ll be asked what file we want to save this to: pressReturnto accept the suggested default ofdraft.txt).
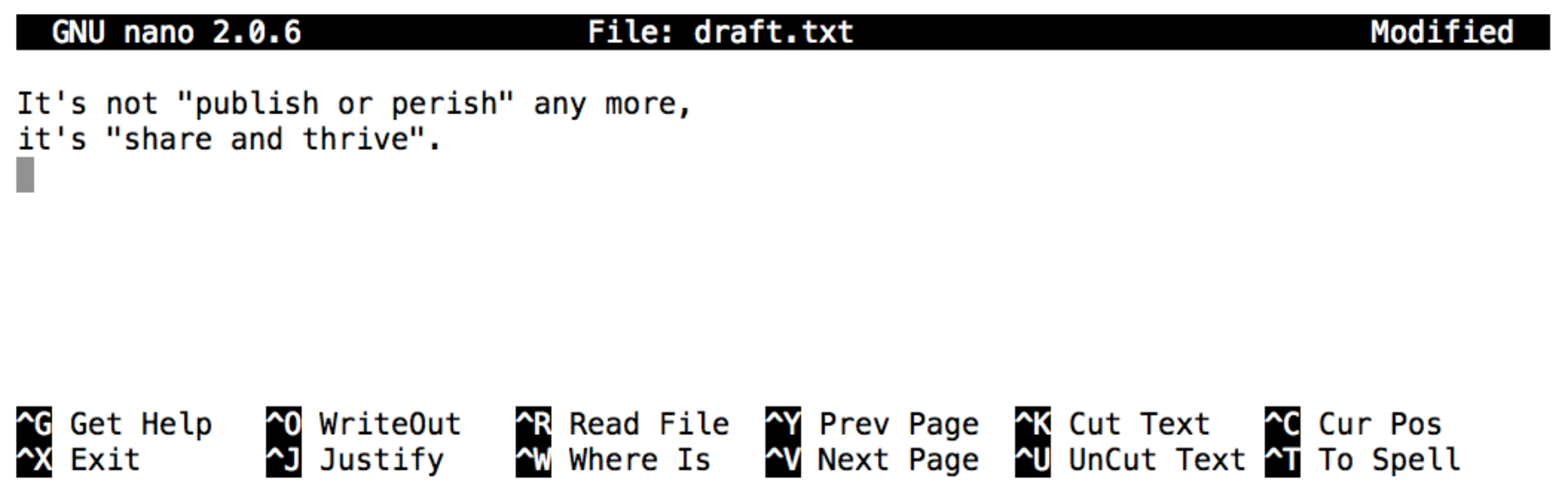
Once our file is saved, we can use
Ctrl+Xto quit the editor and return to the shell.
Control, Ctrl, or ^ Key¶
The Control key is also called the ‘Ctrl’ key. There are various ways in which using the Control key may be described. For example, you may see an instruction to press the
Controlkey and, while holding it down, press theXkey, described as any of:
Control-XControl+XCtrl-XCtrl+X^XC-x
In nano, along the bottom of the screen you’ll see ^G Get Help ^O WriteOut.
This means that you can use Control-G to get help and Control-O to save your
file.
nano doesn’t leave any output on the screen after it exits,
but ls now shows that we have created a file called draft.txt:
$ ls
draft.txt
Creating Files a Different Way¶
We have seen how to create text files using the
nanoeditor. Now, try the following command:
$ touch my_file.txt
What did the
touchcommand do? When you look at your current directory using the GUI file explorer, does the file show up?Use
ls -lto inspect the files. How large ismy_file.txt?When might you want to create a file this way?
Solution
The
touchcommand generates a new file calledmy_file.txtin your current directory. You can observe this newly generated file by typinglsat the command line prompt.my_file.txtcan also be viewed in your GUI file explorer.When you inspect the file with
ls -l, note that the size ofmy_file.txtis 0 bytes. In other words, it contains no data. If you openmy_file.txtusing your text editor it is blank.Some programs do not generate output files themselves, but instead require that empty files have already been generated. When the program is run, it searches for an existing file to populate with its output. The touch command allows you to efficiently generate a blank text file to be used by such programs.
What’s In A Name?¶
You may have noticed that all of Nelle’s files are named ‘something dot something’, and in this part of the lesson, we always used the extension
.txt. This is just a convention: we can call a filemythesisor almost anything else we want. However, most people use two-part names most of the time to help them (and their programs) tell different kinds of files apart. The second part of such a name is called the filename extension, and indicates what type of data the file holds:.txtsignals a plain text file,.cfgis a configuration file full of parameters for some program or other,.pngis a PNG image, and so on.This is just a convention, albeit an important one. Files contain bytes: it’s up to us and our programs to interpret those bytes according to the rules for plain text files, PDF documents, configuration files, images, and so on.
Naming a PNG image of a whale as
whale.mp3doesn’t somehow magically turn it into a recording of whalesong, though it might cause the operating system to try to open it with a music player when someone double-clicks it.
Moving files and directories¶
Returning to the
ag2pi_workshopdirectory,
cd ~/input/ag2pi_workshop
In our thesis directory we have a file draft.txt
which isn’t a particularly informative name,
so let’s change the file’s name using mv,
which is short for ‘move’:
$ mv thesis/draft.txt thesis/quotes.txt
The first argument tells mv what we’re ‘moving’,
while the second is where it’s to go.
In this case,
we’re moving thesis/draft.txt to thesis/quotes.txt,
which has the same effect as renaming the file.
Sure enough,
ls shows us that thesis now contains one file called quotes.txt:
$ ls thesis
quotes.txt
One has to be careful when specifying the target file name, since mv will
silently overwrite any existing file with the same name, which could
lead to data loss. An additional option, mv -i (or mv --interactive),
can be used to make mv ask you for confirmation before overwriting.
Note that mv also works on directories.
Let’s move quotes.txt into the current working directory.
We use mv once again,
but this time we’ll use just the name of a directory as the second argument
to tell mv that we want to keep the filename,
but put the file somewhere new.
(This is why the command is called ‘move’.)
In this case, the directory name we use is the special directory name . that we mentioned earlier.
$ mv thesis/quotes.txt .
The effect is to move the file from the directory it was in to the current working directory.
ls now shows us that thesis is empty:
$ ls thesis
Further, ls with a filename or directory name as an argument only lists that file or directory.
We can use this to see that quotes.txt is still in our current directory:
$ ls quotes.txt
quotes.txt
Moving Files to a new folder¶
After running the following commands, Jamie realizes that she put the files
sucrose.datandmaltose.datinto the wrong folder. The files should have been placed in therawfolder.
$ ls -F
analyzed/ raw/
$ ls -F analyzed
fructose.dat glucose.dat maltose.dat sucrose.dat
$ cd analyzed
Fill in the blanks to move these files to the raw/ folder
(i.e. the one she forgot to put them in)
$ mv sucrose.dat maltose.dat ____/____
Solution
$ mv sucrose.dat maltose.dat ../raw
Recall that .. refers to the parent directory (i.e. one above the current directory)
and that . refers to the current directory.
Copying files and directories¶
The
cpcommand works very much likemv, except it copies a file instead of moving it. We can check that it did the right thing usinglswith two paths as arguments — like most Unix commands,lscan be given multiple paths at once:
$ cp quotes.txt thesis/quotations.txt
$ ls quotes.txt thesis/quotations.txt
quotes.txt thesis/quotations.txt
We can also copy a directory and all its contents by using the
recursive option -r,
e.g. to back up a directory:
$ cp -r thesis thesis_backup
We can check the result by listing the contents of both the thesis and thesis_backup directory:
$ ls thesis thesis_backup
thesis:
quotations.txt
thesis_backup:
quotations.txt
Renaming Files¶
Suppose that you created a plain-text file in your current directory to contain a list of the
statistical tests you will need to do to analyze your data, and named it: statstics.txt
After creating and saving this file you realize you misspelled the filename! You want to correct the mistake, which of the following commands could you use to do so?
cp statstics.txt statistics.txtmv statstics.txt statistics.txtmv statstics.txt .cp statstics.txt .
Solution
No. While this would create a file with the correct name, the incorrectly named file still exists in the directory and would need to be deleted.
Yes, this would work to rename the file.
No, the period(.) indicates where to move the file, but does not provide a new file name; identical file names cannot be created.
No, the period(.) indicates where to copy the file, but does not provide a new file name; identical file names cannot be created.
Moving and Copying¶
What is the output of the closing ls command in the sequence shown below?
$ pwd
/Users/jamie/data
$ ls
proteins.dat
$ mkdir recombined
$ mv proteins.dat recombined/
$ cp recombined/proteins.dat ../proteins-saved.dat
$ ls
proteins-saved.dat recombinedrecombinedproteins.dat recombinedproteins-saved.dat
Solution
We start in the /Users/jamie/data directory, and create a new folder called recombined.
The second line moves (mv) the file proteins.dat to the new folder (recombined).
The third line makes a copy of the file we just moved. The tricky part here is where the file was
copied to. Recall that .. means ‘go up a level’, so the copied file is now in /Users/jamie.
Notice that .. is interpreted with respect to the current working
directory, not with respect to the location of the file being copied.
So, the only thing that will show using ls (in /Users/jamie/data) is the recombined folder.
No, see explanation above.
proteins-saved.datis located at/Users/jamieYes
No, see explanation above.
proteins.datis located at/Users/jamie/data/recombinedNo, see explanation above.
proteins-saved.datis located at/Users/jamie
Removing files and directories¶
Returning to the ag2pi_workshop directory,
let’s tidy up this directory by removing the quotes.txt file we created.
The Unix command we’ll use for this is rm (short for ‘remove’):
$ rm quotes.txt
We can confirm the file has gone using ls:
$ ls quotes.txt
ls: cannot access 'quotes.txt': No such file or directory
Deleting Is Forever¶
`rm` is a powerful command.
The Unix shell doesn’t have a trash bin that we can recover deleted files from (though most graphical interfaces to Unix do). Instead, when we delete files, they are unlinked from the file system so that their storage space on disk can be recycled. Tools for finding and recovering deleted files do exist, but there’s no guarantee they’ll work in any particular situation, since the computer may recycle the file’s disk space right away.
Using rm Safely¶
What happens when we execute rm -i thesis_backup/quotations.txt?
Why would we want this protection when using rm?
$ rm: remove regular file 'thesis_backup/quotations.txt'? y
The -i option will prompt before (every) removal (use Y to confirm deletion or N to keep the file).
The Unix shell doesn’t have a trash bin, so all the files removed will disappear forever.
By using the -i option, we have the chance to check that we are deleting only the files that we want to remove.
If we try to remove the thesis directory using rm thesis,
we get an error message:
$ rm thesis
rm: cannot remove `thesis': Is a directory
This happens because rm by default only works on files, not directories.
rm can remove a directory and all its contents if we use the
recursive option -r, and it will do so without any confirmation prompts:
$ rm -r thesis
Given that there is no way to retrieve files deleted using the shell,
rm -r should be used with great caution (you might consider adding the interactive option rm -r -i).
Operations with multiple files and directories¶
Oftentimes one needs to copy or move several files at once. This can be done by providing a list of individual filenames, or specifying a naming pattern using wildcards.
Copy with Multiple Filenames¶
For this exercise, you can test the commands in the ag2pi_workshop/data directory.
In the example below, what does cp do when given several filenames and a directory name?
$ mkdir backup
$ cp amino-acids.txt animals.txt backup/
In the example below, what does cp do when given three or more file names?
$ ls -F
amino-acids.txt animals.txt backup/ elements/ morse.txt pdb/ planets.txt salmon.txt sunspot.txt
$ cp amino-acids.txt animals.txt morse.txt
Solution
If given more than one file name followed by a directory name (i.e. the destination directory must
be the last argument), cp copies the files to the named directory.
If given three file names, cp throws an error such as the one below, because it is expecting a directory
name as the last argument.
cp: target ‘morse.txt’ is not a directory
Using wildcards for accessing multiple files at once¶
Wildcards¶
*is a wildcard, which matches zero or more characters. Let’s consider theag2pi_workshop/moleculesdirectory:*.pdbmatchesethane.pdb,propane.pdb, and every file that ends with ‘.pdb’. On the other hand,p*.pdbonly matchespentane.pdbandpropane.pdb, because the ‘p’ at the front only matches filenames that begin with the letter ‘p’.
?is also a wildcard, but it matches exactly one character. So?ethane.pdbwould matchmethane.pdbwhereas*ethane.pdbmatches bothethane.pdb, andmethane.pdb.Wildcards can be used in combination with each other e.g.
???ane.pdbmatches three characters followed byane.pdb, givingcubane.pdb ethane.pdb octane.pdb.When the shell sees a wildcard, it expands the wildcard to create a list of matching filenames before running the command that was asked for. As an exception, if a wildcard expression does not match any file, Bash will pass the expression as an argument to the command as it is. For example typing
ls *.pdfin themoleculesdirectory (which contains only files with names ending with.pdb) results in an error message that there is no file calledwcandlssee the lists of file names matching these expressions, but not the wildcards themselves. It is the shell, not the other programs, that deals with expanding wildcards, and this is another example of orthogonal design.
List filenames matching a pattern¶
When run in the
moleculesdirectory, whichlscommand(s) will produce this output?
ethane.pdb methane.pdb
ls *t*ane.pdbls *t?ne.*ls *t??ne.pdbls ethane.*
Solution
The solution is 3.
shows all files whose names contain zero or more characters (
*) followed by the lettert, then zero or more characters (*) followed byane.pdb. This givesethane.pdb methane.pdb octane.pdb pentane.pdb.shows all files whose names start with zero or more characters (
*) followed by the lettert, then a single character (?), thenne.followed by zero or more characters (*). This will give usoctane.pdbandpentane.pdbbut doesn’t match anything which ends inthane.pdb.fixes the problems of option 2 by matching two characters (
??) betweentandne. This is the solution.only shows files starting with
ethane..
More on Wildcards¶
Sam has a directory containing calibration data, datasets, and descriptions of the datasets:
.
├── 2015-10-23-calibration.txt
├── 2015-10-23-dataset1.txt
├── 2015-10-23-dataset2.txt
├── 2015-10-23-dataset_overview.txt
├── 2015-10-26-calibration.txt
├── 2015-10-26-dataset1.txt
├── 2015-10-26-dataset2.txt
├── 2015-10-26-dataset_overview.txt
├── 2015-11-23-calibration.txt
├── 2015-11-23-dataset1.txt
├── 2015-11-23-dataset2.txt
├── 2015-11-23-dataset_overview.txt
├── backup
│ ├── calibration
│ └── datasets
└── send_to_bob
├── all_datasets_created_on_a_23rd
└── all_november_files
Before heading off to another field trip, she wants to back up her data and send some datasets to her colleague Bob. Sam uses the following commands to get the job done:
$ cp *dataset* backup/datasets
$ cp ____calibration____ backup/calibration
$ cp 2015-____-____ send_to_bob/all_november_files/
$ cp ____ send_to_bob/all_datasets_created_on_a_23rd/
Help Sam by filling in the blanks.
The resulting directory structure should look like this
.
├── 2015-10-23-calibration.txt
├── 2015-10-23-dataset1.txt
├── 2015-10-23-dataset2.txt
├── 2015-10-23-dataset_overview.txt
├── 2015-10-26-calibration.txt
├── 2015-10-26-dataset1.txt
├── 2015-10-26-dataset2.txt
├── 2015-10-26-dataset_overview.txt
├── 2015-11-23-calibration.txt
├── 2015-11-23-dataset1.txt
├── 2015-11-23-dataset2.txt
├── 2015-11-23-dataset_overview.txt
├── backup
│ ├── calibration
│ │ ├── 2015-10-23-calibration.txt
│ │ ├── 2015-10-26-calibration.txt
│ │ └── 2015-11-23-calibration.txt
│ └── datasets
│ ├── 2015-10-23-dataset1.txt
│ ├── 2015-10-23-dataset2.txt
│ ├── 2015-10-23-dataset_overview.txt
│ ├── 2015-10-26-dataset1.txt
│ ├── 2015-10-26-dataset2.txt
│ ├── 2015-10-26-dataset_overview.txt
│ ├── 2015-11-23-dataset1.txt
│ ├── 2015-11-23-dataset2.txt
│ └── 2015-11-23-dataset_overview.txt
└── send_to_bob
├── all_datasets_created_on_a_23rd
│ ├── 2015-10-23-dataset1.txt
│ ├── 2015-10-23-dataset2.txt
│ ├── 2015-10-23-dataset_overview.txt
│ ├── 2015-11-23-dataset1.txt
│ ├── 2015-11-23-dataset2.txt
│ └── 2015-11-23-dataset_overview.txt
└── all_november_files
├── 2015-11-23-calibration.txt
├── 2015-11-23-dataset1.txt
├── 2015-11-23-dataset2.txt
└── 2015-11-23-dataset_overview.txt
Solution
$ cp *calibration.txt backup/calibration
$ cp 2015-11-* send_to_bob/all_november_files/
$ cp *-23-dataset* send_to_bob/all_datasets_created_on_a_23rd/
Organizing Directories and Files¶
Jamie is working on a project and she sees that her files aren’t very well organized:
$ ls -F
analyzed/ fructose.dat raw/ sucrose.dat
The fructose.dat and sucrose.dat files contain output from her data
analysis. What command(s) covered in this lesson does she need to run so that the commands below will
produce the output shown?
$ ls -F
analyzed/ raw/
$ ls analyzed
fructose.dat sucrose.dat
Solution
mv *.dat analyzed
Jamie needs to move her files fructose.dat and sucrose.dat to the analyzed directory.
The shell will expand *.dat to match all .dat files in the current directory.
The mv command then moves the list of .dat files to the ‘analyzed’ directory.
Reproduce a folder structure¶
You’re starting a new experiment, and would like to duplicate the directory structure from your previous experiment so you can add new data.
Assume that the previous experiment is in a folder called ‘2016-05-18’,
which contains a data folder that in turn contains folders named raw and
processed that contain data files. The goal is to copy the folder structure
of the 2016-05-18-data folder into a folder called 2016-05-20
so that your final directory structure looks like this:
2016-05-20/
└── data
├── processed
└── raw
Which of the following set of commands would achieve this objective? What would the other commands do?
$ mkdir 2016-05-20
$ mkdir 2016-05-20/data
$ mkdir 2016-05-20/data/processed
$ mkdir 2016-05-20/data/raw
$ mkdir 2016-05-20
$ cd 2016-05-20
$ mkdir data
$ cd data
$ mkdir raw processed
$ mkdir 2016-05-20/data/raw
$ mkdir 2016-05-20/data/processed
$ mkdir -p 2016-05-20/data/raw
$ mkdir -p 2016-05-20/data/processed
$ mkdir 2016-05-20
$ cd 2016-05-20
$ mkdir data
$ mkdir raw processed
Solution
The first two sets of commands achieve this objective. The first set uses relative paths to create the top level directory before the subdirectories. The third set of commands will give an error because the default behavior of ``mkdir`` won't create a subdirectory of a non-existant directory: the intermediate level folders must be created first. The fourth set of commands achieve this objective. Remember, the ``-p`` option, followed by a path of one or more directories, will cause ``mkdir`` to create any intermediate subdirectories as required. The final set of commands generates the 'raw' and 'processed' directories at the same level as the 'data' directory.Pipes and Filters¶
Questions:
“How can I combine existing commands to do new things?”
Objectives:
Redirect a command’s output to a file.
Process a file instead of keyboard input using redirection.
Construct command pipelines with two or more stages.
Explain what usually happens if a program or pipeline isn’t given any input to process.
Explain Unix’s ‘small pieces, loosely joined’ philosophy.
Keypoints:
catdisplays the contents of its inputs.headdisplays the first 10 lines of its input.taildisplays the last 10 lines of its input.sortsorts its inputs.wccounts lines, words, and characters in its inputs.command [file]redirects a command’s output to a file (overwriting any existing content).command >[file]appends a command’s output to a file.[first] | [second]is a pipeline: the output of the first command is used as the input to the second.The best way to use the shell is to use pipes to combine simple single-purpose programs (filters).
Now that we know a few basic commands,
we can finally look at the shell’s most powerful feature:
the ease with which it lets us combine existing programs in new ways.
We’ll start with the directory called ag2pi_workshop/molecules
that contains six files describing some simple organic molecules.
The .pdb extension indicates that these files are in Protein Data Bank format,
a simple text format that specifies the type and position of each atom in the molecule.
$ ls molecules
cubane.pdb ethane.pdb methane.pdb
octane.pdb pentane.pdb propane.pdb
Let’s go into that directory with cd and run an example command wc cubane.pdb:
$ cd molecules
$ wc cubane.pdb
20 156 1158 cubane.pdb
wc is the ‘word count’ command:
it counts the number of lines, words, and characters in files (from left to right, in that order).
If we run the command wc *.pdb, the * in *.pdb matches zero or more characters,
so the shell turns *.pdb into a list of all .pdb files in the current directory:
$ wc *.pdb
20 156 1158 cubane.pdb
12 84 622 ethane.pdb
9 57 422 methane.pdb
30 246 1828 octane.pdb
21 165 1226 pentane.pdb
15 111 825 propane.pdb
107 819 6081 total
Note that wc *.pdb also shows the total number of all lines in the last line of the output.
If we run wc -l instead of just wc,
the output shows only the number of lines per file:
$ wc -l *.pdb
20 cubane.pdb
12 ethane.pdb
9 methane.pdb
30 octane.pdb
21 pentane.pdb
15 propane.pdb
107 total
The -m and -w options can also be used with the wc command, to show
only the number of characters or the number of words in the files.
What happens if a command is supposed to process a file, but we don’t give it a filename? For example, what if we type:
$ wc -l
but don’t type *.pdb (or anything else) after the command?
Since it doesn’t have any filenames, wc assumes it is supposed to
process input given at the command prompt, so it just sits there and waits for us to give
it some data interactively. From the outside, though, all we see is it
sitting there: the command doesn’t appear to do anything.
If you make this kind of mistake, you can escape out of this state by holding down
the control key Ctrl and typing the letter C``once and letting go of the ``Ctrl key.
Ctrl + C
Which of these files contains the fewest lines? It’s an easy question to answer when there are only six files, but what if there were 6000? Our first step toward a solution is to run the command:
$ wc -l *.pdb > lengths.txt
The greater than symbol, >, tells the shell to redirect the command’s output
to a file instead of printing it to the screen. (This is why there is no screen output:
everything that wc would have printed has gone into the
file lengths.txt instead.) The shell will create
the file if it doesn’t exist. If the file exists, it will be
silently overwritten, which may lead to data loss and thus requires
some caution.
ls lengths.txt confirms that the file exists:
$ ls lengths.txt
lengths.txt
We can now send the content of lengths.txt to the screen using cat lengths.txt.
The cat command gets its name from ‘concatenate’ i.e. join together,
and it prints the contents of files one after another.
There’s only one file in this case,
so cat just shows us what it contains:
$ cat lengths.txt
20 cubane.pdb
12 ethane.pdb
9 methane.pdb
30 octane.pdb
21 pentane.pdb
15 propane.pdb
107 total
We’ll continue to use cat in this lesson, for convenience and consistency,
but it has the disadvantage that it always dumps the whole file onto your screen.
More useful in practice is the command less,
which you use with less lengths.txt.
This displays a screenful of the file, and then stops.
You can go forward one screenful by pressing the spacebar,
or back one by pressing b. Press q to quit.
Now let’s use the sort command to sort its contents.
If we run sort on a file containing the following lines:
10
2
19
22
6
the output is:
10
19
2
22
6
If we run sort -n on the same input, we get this instead:
2
6
10
19
22
Explain why -n has this effect.
Solution
The -n option specifies a numerical rather than an alphanumerical sort.
We will also use the -n option to specify that the sort is
numerical instead of alphanumerical.
This does not change the file;
instead, it sends the sorted result to the screen:
$ sort -n lengths.txt
9 methane.pdb
12 ethane.pdb
15 propane.pdb
20 cubane.pdb
21 pentane.pdb
30 octane.pdb
107 total
We can put the sorted list of lines in another temporary file called sorted-lengths.txt
by putting sorted-lengths.txt after the command,
just as we used lengths.txt to put the output of wc into lengths.txt.
Once we’ve done that,
we can run another command called head to get the first few lines in sorted-lengths.txt:
$ sort -n lengths.txt > sorted-lengths.txt
$ head -n 1 sorted-lengths.txt
9 methane.pdb
Using -n 1 with head tells it that
we only want the first line of the file;
-n 20 would get the first 20, and so on.
Since sorted-lengths.txt contains the lengths of our files ordered from least to greatest,
the output of head must be the file with the fewest lines.
It’s a very bad idea to try redirecting the output of a command that operates on a file to the same file. For example:
$ sort -n lengths.txt > lengths.txt
Doing something like this may give you
incorrect results and/or delete
the contents of lengths.txt.
We have seen the use of >, but there is a similar operator >> which works slightly differently.
We’ll learn about the differences between these two operators by printing some strings.
We can use the echo command to print strings of text e.g.
$ echo The echo command prints text
The echo command prints text
Now test the commands below to reveal the difference between the two operators:
$ echo hello > testfile01.txt
and:
$ echo hello >> testfile02.txt
Hint: Try executing each command twice in a row and then examining the output files.
In the first example with >, the string ‘hello’ is written to testfile01.txt,
but the file gets overwritten each time we run the command.
We see from the second example that the >> operator also writes ‘hello’ to a file
(in this case testfile02.txt),
but appends the string to the file if it already exists (i.e. when we run it for the second time).
We have already met the head command, which prints lines from the start of a file.
tail is similar, but prints lines from the end of a file instead.
Consider the file ag2pi_workshop/data/animals.txt.
After these commands, select the answer that
corresponds to the file animals-subset.txt:
$ head -n 3 animals.txt > animals-subset.txt
$ tail -n 2 animals.txt > animals-subset.txt
The first three lines of
animals.txtThe last two lines of
animals.txtThe first three lines and the last two lines of
animals.txtThe second and third lines of
animals.txt
If you think this is confusing, you’re in good company:
even once you understand what wc, sort, and head do,
all those intermediate files make it hard to follow what’s going on.
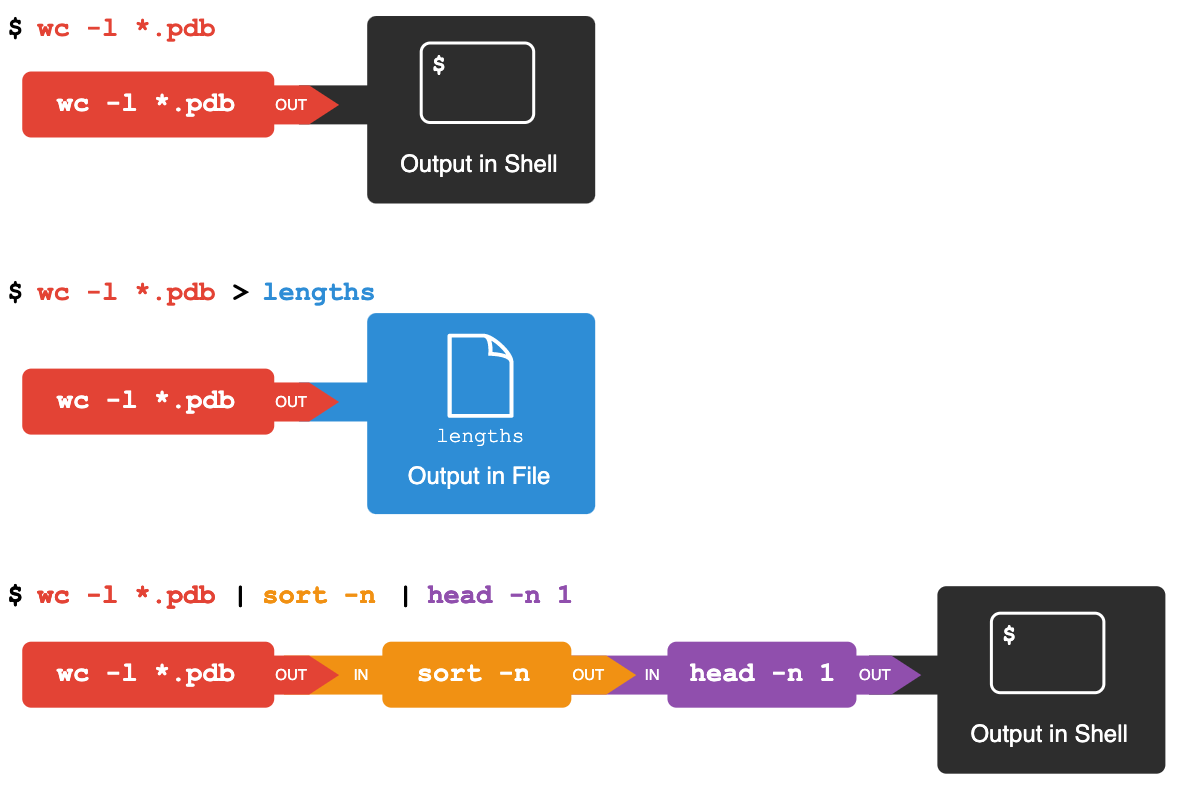
We can make it easier to understand by running sort and head together:
$ sort -n lengths.txt | head -n 1
9 methane.pdb
The vertical bar, |, between the two commands is called a pipe.
It tells the shell that we want to use
the output of the command on the left
as the input to the command on the right.
Nothing prevents us from chaining pipes consecutively.
That is, we can for example send the output of wc directly to sort,
and then the resulting output to head.
Thus we first use a pipe to send the output of wc to sort:
$ wc -l *.pdb | sort -n
9 methane.pdb
12 ethane.pdb
15 propane.pdb
20 cubane.pdb
21 pentane.pdb
30 octane.pdb
107 total
And now we send the output of this pipe, through another pipe, to head, so that the full pipeline becomes:
$ wc -l *.pdb | sort -n | head -n 1
9 methane.pdb
This is exactly like a mathematician nesting functions like log(3x)
and saying ‘the log of three times x’.
In our case,
the calculation is ‘head of sort of line count of *.pdb’.
The redirection and pipes used in the last few commands are illustrated below:
In our current directory, we want to find the 3 files which have the least number of lines. Which command listed below would work?
wc -l * sort -n head -n 3wc -l * | sort -n | head -n 1-3wc -l * | head -n 3 | sort -nwc -l * | sort -n | head -n 3
This idea of linking programs together is why Unix has been so successful.
Instead of creating enormous programs that try to do many different things,
Unix programmers focus on creating lots of simple tools that each do one job well,
and that work well with each other.
This programming model is called ‘pipes and filters’.
We’ve already seen pipes;
a filter is a program like wc or sort
that transforms a stream of input into a stream of output.
Almost all of the standard Unix tools can work this way:
unless told to do otherwise,
they read from standard input,
do something with what they’ve read,
and write to standard output.
The key is that any program that reads lines of text from standard input and writes lines of text to standard output can be combined with every other program that behaves this way as well. You can and should write your programs this way so that you and other people can put those programs into pipes to multiply their power.
A file called animals.txt (in the ag2pi_workshop/data folder) contains the following data:
2012-11-05,deer
2012-11-05,rabbit
2012-11-05,raccoon
2012-11-06,rabbit
2012-11-06,deer
2012-11-06,fox
2012-11-07,rabbit
2012-11-07,bear
What text passes through each of the pipes and the final redirect in the pipeline below?
$ cat animals.txt | head -n 5 | tail -n 3 | sort -r > final.txt
Hint: build the pipeline up one command at a time to test your understanding
The head command extracts the first 5 lines from animals.txt.
Then, the last 3 lines are extracted from the previous 5 by using the tail command.
With the sort -r command those 3 lines are sorted in reverse order and finally,
the output is redirected to a file final.txt.
The content of this file can be checked by executing cat final.txt.
The file should contain the following lines:
2012-11-06,rabbit
2012-11-06,deer
2012-11-05,raccoon
For the file animals.txt from the previous exercise, consider the following command:
$ cut -d , -f 2 animals.txt
The cut command is used to remove or ‘cut out’ certain sections of each line in the file,
and cut expects the lines to be separated into columns by a Tab character.
A character used in this way is a called a delimiter.
In the example above we use the -d option to specify the comma as our delimiter character.
We have also used the -f option to specify that we want to extract the second field (column).
This gives the following output:
deer
rabbit
raccoon
rabbit
deer
fox
rabbit
bear
The uniq command filters out adjacent matching lines in a file.
How could you extend this pipeline (using uniq and another command) to find
out what animals the file contains (without any duplicates in their
names)?
$ cut -d , -f 2 animals.txt | sort | uniq
The file animals.txt contains 8 lines of data formatted as follows:
2012-11-05,deer
2012-11-05,rabbit
2012-11-05,raccoon
2012-11-06,rabbit
...
The uniq command has a -c option which gives a count of the
number of times a line occurs in its input. Assuming your current
directory is ag2pi_workshop/data/, what command would you use to produce
a table that shows the total count of each type of animal in the file?
sort animals.txt | uniq -csort -t, -k2,2 animals.txt | uniq -ccut -d, -f 2 animals.txt | uniq -ccut -d, -f 2 animals.txt | sort | uniq -ccut -d, -f 2 animals.txt | sort | uniq -c | wc -l
Option 4. is the correct answer.
If you have difficulty understanding why, try running the commands, or sub-sections of
the pipelines (make sure you are in the ag2pi_workshop/data directory).
Nelle has run her samples through the assay machines
and created 17 files in the north-pacific-gyre/2012-07-03 directory described earlier.
As a quick check, starting from her home directory, Nelle types:
$ cd north-pacific-gyre/2012-07-03
$ wc -l *.txt
The output is 18 lines that look like this:
300 NENE01729A.txt
300 NENE01729B.txt
300 NENE01736A.txt
300 NENE01751A.txt
300 NENE01751B.txt
300 NENE01812A.txt
... ...
Now she types this:
$ wc -l *.txt | sort -n | head -n 5
240 NENE02018B.txt
300 NENE01729A.txt
300 NENE01729B.txt
300 NENE01736A.txt
300 NENE01751A.txt
Whoops: one of the files is 60 lines shorter than the others. When she goes back and checks it, she sees that she did that assay at 8:00 on a Monday morning — someone was probably in using the machine on the weekend, and she forgot to reset it. Before re-running that sample, she checks to see if any files have too much data:
$ wc -l *.txt | sort -n | tail -n 5
300 NENE02040B.txt
300 NENE02040Z.txt
300 NENE02043A.txt
300 NENE02043B.txt
5040 total
Those numbers look good — but what’s that ‘Z’ doing there in the third-to-last line? All of her samples should be marked ‘A’ or ‘B’; by convention, her lab uses ‘Z’ to indicate samples with missing information. To find others like it, she does this:
$ ls *Z.txt
NENE01971Z.txt NENE02040Z.txt
Sure enough,
when she checks the log on her laptop,
there’s no depth recorded for either of those samples.
Since it’s too late to get the information any other way,
she must exclude those two files from her analysis.
She could delete them using rm,
but there are actually some analyses she might do later where depth doesn’t matter,
so instead, she’ll have to be careful later on to select files using the wildcard expression *[AB].txt.
As always, the * matches any number of characters;
the expression [AB] matches either an ‘A’ or a ‘B’,
so this matches all the valid data files she has.
Wildcard expressions can be very complex, but you can sometimes write
them in ways that only use simple syntax, at the expense of being a bit more verbose.
Consider the directory ag2pi_workshop/north-pacific-gyre/2012-07-03 :
the wildcard expression *[AB].txt
matches all files ending in A.txt or B.txt. Imagine you forgot about
this.
Can you match the same set of files with basic wildcard expressions that do not use the
[]syntax? Hint: You may need more than one command, or two arguments to thelscommand.If you used two commands, the files in your output will match the same set of files in this example. What is the small difference between the outputs?
If you used two commands, under what circumstances would your new expression produce an error message where the original one would not?
A solution using two wildcard commands:
$ ls *A.txt $ ls *B.txt
A solution using one command but with two arguments:
$ ls *A.txt *B.txt
The output from the two new commands is separated because there are two commands.
When there are no files ending in
A.txt, or there are no files ending inB.txt, then one of the two commands will fail.
Suppose you want to delete your processed data files, and only keep
your raw files and processing script to save storage.
The raw files end in .dat and the processed files end in .txt.
Which of the following would remove all the processed data files,
and only the processed data files?
rm ?.txtrm *.txtrm * .txtrm *.*
This would remove
.txtfiles with one-character namesThis is correct answer
The shell would expand
*to match everything in the current directory, so the command would try to remove all matched files and an additional file called.txtThe shell would expand
*.*to match all files with any extension, so this command would delete all files
Finding Things¶
Questions:
How can I find files?
How can I find things in files?
Objectives:
Use
grepto select lines from text files that match simple patterns.Use
findto find files and directories whose names match simple patterns.Use the output of one command as the command-line argument(s) to another command.
Explain what is meant by ‘text’ and ‘binary’ files, and why many common tools don’t handle the latter well.
Keypoints:
findfinds files with specific properties that match patterns.grepselects lines in files that match patterns.--helpis an option supported by many bash commands, and programs that can be run from within Bash, to display more information on how to use these commands or programs.man [command]displays the manual page for a given command.$([command])inserts a command’s output in place.
In the same way that many of us now use ‘Google’ as a verb meaning ‘to find’, Unix programmers often use the word ‘grep’. ‘grep’ is a contraction of ‘global/regular expression/print’, a common sequence of operations in early Unix text editors. It is also the name of a very useful command-line program.
grep finds and prints lines in files that match a pattern.
For our examples,
we will use a file that contains three haikus taken from a
1998 competition in Salon magazine. For this set of examples,
we’re going to be working in the writing subdirectory:
$ cd
$ cd input/ag2pi_workshop/writing
$ cat haiku.txt
The Tao that is seen
Is not the true Tao, until
You bring fresh toner.
With searching comes loss
and the presence of absence:
"My Thesis" not found.
Yesterday it worked
Today it is not working
Software is like that.
We haven’t linked to the original haikus because they don’t appear to be on Salon’s site any longer. As Jeff Rothenberg said, ‘Digital information lasts forever — or five years, whichever comes first.’ Luckily, popular content often has backups.
Let’s find lines that contain the word ‘not’:
$ grep not haiku.txt
Is not the true Tao, until
"My Thesis" not found
Today it is not working
Here, not is the pattern we’re searching for. The grep command searches through the file, looking for matches to the pattern specified. To use it type grep, then the pattern we’re searching for and finally the name of the file (or files) we’re searching in.
The output is the three lines in the file that contain the letters ‘not’.
By default, grep searches for a pattern in a case-sensitive way. In addition, the search pattern we have selected does not have to form a complete word, as we will see in the next example.
Let’s search for the pattern: ‘The’.
$ grep The haiku.txt
The Tao that is seen
"My Thesis" not found.
This time, two lines that include the letters ‘The’ are outputted, one of which contained our search pattern within a larger word, ‘Thesis’.
To restrict matches to lines containing the word ‘The’ on its own,
we can give grep with the -w option.
This will limit matches to word boundaries.
Later in this lesson, we will also see how we can change the search behavior of grep with respect to its case sensitivity.
$ grep -w The haiku.txt
The Tao that is seen
Note that a ‘word boundary’ includes the start and end of a line, so not
just letters surrounded by spaces.
Sometimes we don’t
want to search for a single word, but a phrase. This is also easy to do with
grep by putting the phrase in quotes.
$ grep -w "is not" haiku.txt
Today it is not working
We’ve now seen that you don’t have to have quotes around single words, but it is useful to use quotes when searching for multiple words. It also helps to make it easier to distinguish between the search term or phrase and the file being searched. We will use quotes in the remaining examples.
Another useful option is -n, which numbers the lines that match:
$ grep -n "it" haiku.txt
5:With searching comes loss
9:Yesterday it worked
10:Today it is not working
Here, we can see that lines 5, 9, and 10 contain the letters ‘it’.
We can combine options (i.e. flags) as we do with other Unix commands.
For example, let’s find the lines that contain the word ‘the’. We can combine
the option -w to find the lines that contain the word ‘the’ and -n to number the lines that match:
$ grep -n -w "the" haiku.txt
2:Is not the true Tao, until
6:and the presence of absence:
Now we want to use the option -i to make our search case-insensitive:
$ grep -n -w -i "the" haiku.txt
1:The Tao that is seen
2:Is not the true Tao, until
6:and the presence of absence:
Now, we want to use the option -v to invert our search, i.e., we want to output
the lines that do not contain the word ‘the’.
$ grep -n -w -v "the" haiku.txt
1:The Tao that is seen
3:You bring fresh toner.
4:
5:With searching comes loss
7:"My Thesis" not found.
8:
9:Yesterday it worked
10:Today it is not working
11:Software is like that.
If we use the -r (recursive) option,
grep can search for a pattern recursively through a set of files in subdirectories.
Let’s search recursively for Yesterday in the ag2pi_workshop/writing directory:
grep -r Yesterday .
data/LittleWomen.txt:"Yesterday, when Aunt was asleep and I was trying to be as still as a
data/LittleWomen.txt:Yesterday at dinner, when an Austrian officer stared at us and then
data/LittleWomen.txt:Yesterday was a quiet day spent in teaching, sewing, and writing in my
haiku.txt:Yesterday it worked
grep has lots of other options. To find out what they are, we can type:
$ grep --help
Usage: grep [OPTION]... PATTERN [FILE]...
Search for PATTERN in each FILE or standard input.
PATTERN is, by default, a basic regular expression (BRE).
Example: grep -i 'hello world' menu.h main.c
Regexp selection and interpretation:
-E, --extended-regexp PATTERN is an extended regular expression (ERE)
-F, --fixed-strings PATTERN is a set of newline-separated fixed strings
-G, --basic-regexp PATTERN is a basic regular expression (BRE)
-P, --perl-regexp PATTERN is a Perl regular expression
-e, --regexp=PATTERN use PATTERN for matching
-f, --file=FILE obtain PATTERN from FILE
-i, --ignore-case ignore case distinctions
-w, --word-regexp force PATTERN to match only whole words
-x, --line-regexp force PATTERN to match only whole lines
-z, --null-data a data line ends in 0 byte, not newline
Miscellaneous:
... ... ...
Which command would result in the following output:
and the presence of absence:
grep "of" haiku.txtgrep -E "of" haiku.txtgrep -w "of" haiku.txtgrep -i "of" haiku.txt
The correct answer is 3, because the -w option looks only for whole-word matches.
The other options will also match ‘of’ when part of another word.
grep’s real power doesn’t come from its options, though; it comes from
the fact that patterns can include wildcards. (The technical name for
these is regular expressions, which
is what the ‘re’ in ‘grep’ stands for.) Regular expressions are both complex
and powerful; if you want to do complex searches, please look at the lesson
on our website. As a taster, we can
find lines that have an ‘o’ in the second position like this:
$ grep -E "^.o" haiku.txt
You bring fresh toner.
Today it is not working
Software is like that.
We use the -E option and put the pattern in quotes to prevent the shell
from trying to interpret it. (If the pattern contained a *, for
example, the shell would try to expand it before running grep.) The
^ in the pattern anchors the match to the start of the line. The .
matches a single character (just like ? in the shell), while the o
matches an actual ‘o’.
Leah has several hundred data files saved in one directory, each of which is formatted like this:
2013-11-05,deer,5
2013-11-05,rabbit,22
2013-11-05,raccoon,7
2013-11-06,rabbit,19
2013-11-06,deer,2
She wants to write a shell script that takes a species as the first command-line argument
and a directory as the second argument. The script should return one file called species.txt
containing a list of dates and the number of that species seen on each date.
For example using the data shown above, rabbit.txt would contain:
2013-11-05,22
2013-11-06,19
Put these commands and pipes in the right order to achieve this:
cut -d : -f 2
|
grep -w $1 -r $2
|
$1.txt
cut -d , -f 1,3
Hint: use man grep to look for how to grep text recursively in a directory
and man cut to select more than one field in a line.
An example of such a file is provided in ag2pi_workshop/data/animal-counts/animals.txt
grep -w $1 -r $2 | cut -d : -f 2 | cut -d , -f 1,3 $1.txt
You would call the script above like this:
$ bash count-species.sh bear .
While grep finds lines in files,
the find command finds files themselves.
Again,
it has a lot of options;
to show how the simplest ones work, we’ll use the directory tree shown below.
Nelle’s writing directory contains one file called haiku.txt and three subdirectories:
thesis (which contains a sadly empty file, empty-draft.md);
data (which contains three files LittleWomen.txt, one.txt and two.txt);
and a tools directory that contains the programs format and stats,
and a subdirectory called old, with a file oldtool.
For our first command,
let’s run find . (remember to run this command from the ag2pi_workshop/writing folder).
$ find .
.
./data
./data/one.txt
./data/LittleWomen.txt
./data/two.txt
./tools
./tools/format
./tools/old
./tools/old/oldtool
./tools/stats
./haiku.txt
./thesis
./thesis/empty-draft.md
As always,
the . on its own means the current working directory,
which is where we want our search to start.
find’s output is the names of every file and directory
under the current working directory.
This can seem useless at first but find has many options
to filter the output and in this lesson we will discover some
of them.
The first option in our list is
-type d that means ‘things that are directories’.
Sure enough,
find’s output is the names of the five directories in our little tree
(including .):
$ find . -type d
./
./data
./thesis
./tools
./tools/old
Notice that the objects find finds are not listed in any particular order.
If we change -type d to -type f,
we get a listing of all the files instead:
$ find . -type f
./haiku.txt
./tools/stats
./tools/old/oldtool
./tools/format
./thesis/empty-draft.md
./data/one.txt
./data/LittleWomen.txt
./data/two.txt
Now let’s try matching by name:
$ find . -name *.txt
./haiku.txt
We expected it to find all the text files,
but it only prints out ./haiku.txt.
The problem is that the shell expands wildcard characters like * before commands run.
Since *.txt in the current directory expands to haiku.txt,
the command we actually ran was:
$ find . -name haiku.txt
find did what we asked; we just asked for the wrong thing.
To get what we want,
let’s do what we did with grep:
put *.txt in quotes to prevent the shell from expanding the * wildcard.
This way,
find actually gets the pattern *.txt, not the expanded filename haiku.txt:
$ find . -name "*.txt"
./data/one.txt
./data/LittleWomen.txt
./data/two.txt
./haiku.txt
ls and find can be made to do similar things given the right options,
but under normal circumstances,
ls lists everything it can,
while find searches for things with certain properties and shows them.
As we said earlier,
the command line’s power lies in combining tools.
We’ve seen how to do that with pipes;
let’s look at another technique.
As we just saw,
find . -name "*.txt" gives us a list of all text files in or below the current directory.
How can we combine that with wc -l to count the lines in all those files?
The simplest way is to put the find command inside $():
$ wc -l $(find . -name "*.txt")
11 ./haiku.txt
300 ./data/two.txt
21022 ./data/LittleWomen.txt
70 ./data/one.txt
21403 total
When the shell executes this command,
the first thing it does is run whatever is inside the $().
It then replaces the $() expression with that command’s output.
Since the output of find is the four filenames ./data/one.txt, ./data/LittleWomen.txt, ./data/two.txt, and ./haiku.txt,
the shell constructs the command:
$ wc -l ./data/one.txt ./data/LittleWomen.txt ./data/two.txt ./haiku.txt
which is what we wanted.
This expansion is exactly what the shell does when it expands wildcards like * and ?,
but lets us use any command we want as our own ‘wildcard’.
It’s very common to use find and grep together.
The first finds files that match a pattern;
the second looks for lines inside those files that match another pattern.
Here, for example, we can find PDB files that contain iron atoms
by looking for the string ‘FE’ in all the .pdb files above the current directory:
$ grep "FE" $(find .. -name "*.pdb")
../data/pdb/heme.pdb:ATOM 25 FE 1 -0.924 0.535 -0.518
The -v option to grep inverts pattern matching, so that only lines
which do not match the pattern are printed. Given that, which of
the following commands will find all files in /data whose names
end in s.txt but whose names also do not contain the string net?
(For example, animals.txt or amino-acids.txt but not planets.txt.)
Once you have thought about your answer, you can test the commands in the ag2pi_workshop
directory.
find data -name "*s.txt" | grep -v netfind data -name *s.txt | grep -v netgrep -v "net" $(find data -name "*s.txt")None of the above.
The correct answer is 1. Putting the match expression in quotes prevents the shell
expanding it, so it gets passed to the find command.
Option 2 is incorrect because the shell expands *s.txt instead of passing the wildcard
expression to find.
Option 3 is incorrect because it searches the contents of the files for lines which do not match ‘net’, rather than searching the file names.
We have focused exclusively on finding patterns in text files. What if your data is stored as images, in databases, or in some other format?
A handful of tools extend grep to handle a few non text formats. But a
more generalizable approach is to convert the data to text, or
extract the text-like elements from the data. On the one hand, it makes simple
things easy to do. On the other hand, complex things are usually impossible. For
example, it’s easy enough to write a program that will extract X and Y
dimensions from image files for grep to play with, but how would you
write something to find values in a spreadsheet whose cells contained
formulas?
A last option is to recognize that the shell and text processing have their limits, and to use another programming language. When the time comes to do this, don’t be too hard on the shell: many modern programming languages have borrowed a lot of ideas from it, and imitation is also the sincerest form of praise.
The Unix shell is older than most of the people who use it. It has survived so long because it is one of the most productive programming environments ever created — maybe even the most productive. Its syntax may be cryptic, but people who have mastered it can experiment with different commands interactively, then use what they have learned to automate their work. Graphical user interfaces may be better at the first, but the shell is still unbeaten at the second. And as Alfred North Whitehead wrote in 1911, ‘Civilization advances by extending the number of important operations which we can perform without thinking about them.’
Write a short explanatory comment for the following shell script:
wc -l $(find . -name "*.dat") | sort -n
Find all files with a
.datextension recursively from the current directoryCount the number of lines each of these files contains
Sort the output from step 2. numerically
- Lesson content modified from The Carpentries: The Unix Shell
- Under The Carpentries License:
Fix or improve this documentation
- Search for an answer:
- Ask us for help:
click
 on the lower right-hand side of the page
on the lower right-hand side of the page
- Report an issue or submit a change:
Send feedback: learning@CyVerse.org